Tools
SNPs "Single Nucleotide Polymorphism"
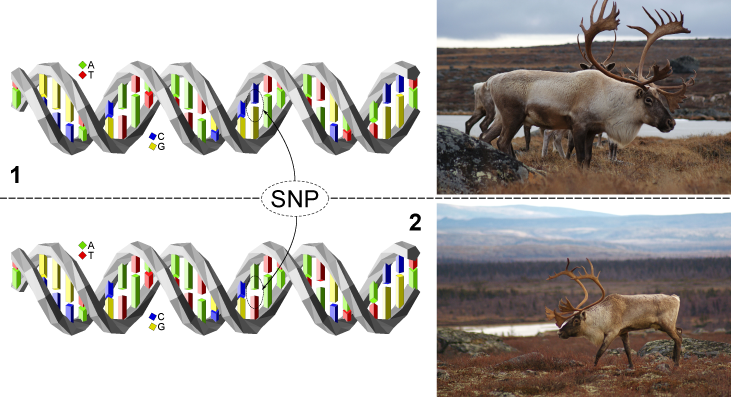
A SNP refers to a location in a DNA sequence where there is a single letter difference between two DNA strands. In other words, we are all different mainly because of differences in our DNA sequence. In mammals, the reproduction process involves copulation between a male and a female individual. Since the two individuals are different, the paternal DNA sequence contains differences compared to the sequence inherited from the mother. We thus carry two DNA strands that are nearly identical except for certain differences that make us unique. There are multiple causes for these differences, because every imaginable modification that can occur in a letter sequence is present in every genome. Hence, letters can be added, removed, or substituted for another.
SNPs are an example of the latter type where a letter is replaced by another. While it is possible to modify multiple consecutive letters within a sequence, SNP modifications refer to cases where a single letter is modified.
Mammalian genomes have fairly similar sizes of approximately 3 billion letters. SNPs occur relatively frequently, and every genome can possibly harbor about 10 million such modifications given a gross estimate of one SNP for every 300 letters on average. This is only an estimate, but the large number of possible SNPs implies that they are scattered all across the genome. Since these differences are inherited from the parents, the characterization of an individual's SNPs gives a description of that individual's genome which can then be compared to that of other individuals. This allows, for instance, to identify families within herds, establish kinship, and determine the level of genetic variability among and between groups.
This information is important because typically, a population is healthy when several families contribute to the progeny, and more extensive genetic diversity allows a population to better adapt to any type of changes. Conversely, a very homogeneous population is generally more vulnerable to stress.
SNP chip
SNP chip technology is used to interrogate a genome to determine what letter appears at a specific location. Genotyping is the process of identifying a single letter in hundreds to hundreds of thousands of regions across the genome. The basic principle of genotyping initially requires knowing where the SNPs to interrogate are found and what letters could possibly be present. While different technological variations are available through different suppliers, the general concept involves the immobilization on a solid-state surface of a tiny DNA fragment corresponding to the sequence of letters found adjacent to the location of the SNP to be analyzed. The individual's DNA is cut in small fragments, and through pairing with the immobilized letter sequence, the complementary fragment will bond to form a double strand of DNA. The four possible letters (A, G, T, and C) are then added individually to determine which one appears at the end of the immobilized strand. Each of the four letters carries a distinct fluorescent molecule. The letter that was added is determined by detecting the colour emitted. Each individual carries two DNA strands – one inherited from the father and the other from the mother; therefore, the maximum number of colours that can possibly be detected for a specific SNP for each individual is two. Miniaturization of the process now makes it possible to interrogate nearly 1.2 million SNPs on a surface the size of a microscope slide (2.5 cm x 7.5 cm). With the tool we are currently developing for the caribou/reindeer, the chip will allow 60,000 SNPs to be interrogated. These are distributed throughout the genome to provide information across the length of the DNA molecule. |  |
